|
Title: Returning to the roots: resolution, reproducibility, and robusticity in the phylogenetic inference of Dissorophidae (Amphibia: Temnospondyli) Authors: B.M. Gee Journal: PeerJ DOI: 10.7717/peerj.12423 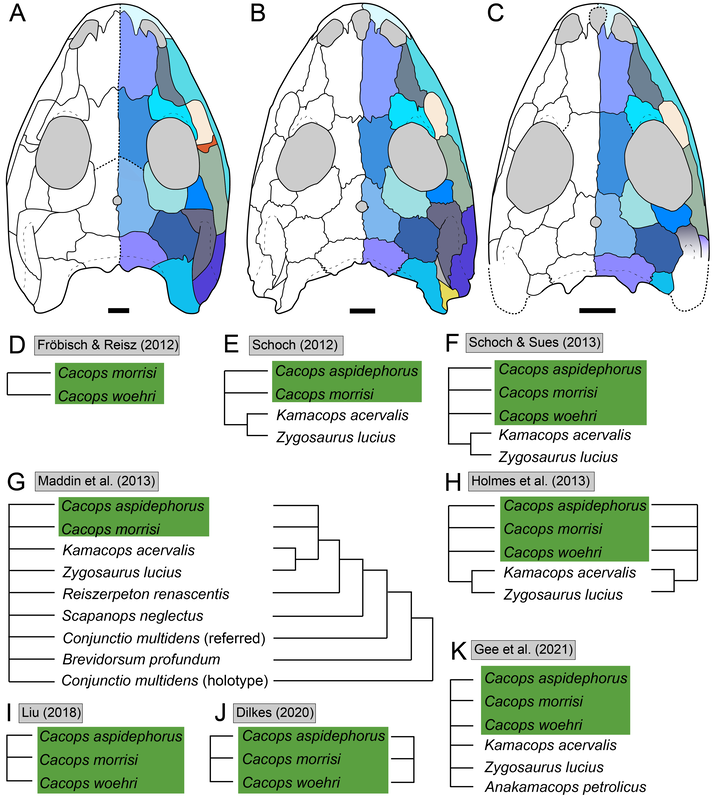 General summary: Phylogenetics, the means of inferring relationships among organisms and one way of classifying them, is a timeless part of paleontology, in part because our datasets are much more limited than those for modern organisms in which genetic barcoding is both widely accessible and robustly informative. As a result, there is often a lot more uncertainty and disparity among different analyses, and people should generally be skeptical when someone says there is a strong consensus for a given group, no matter how well-known (paleo nerds will definitely recall the debate a few years back about what 'Dinosauria' constituted). Temnospondyls are generally not well-known, and this means that they are not as well-studied. This often has the effect of resulting in one or two people producing a lot of the work for a given group, which can lead to an overstated consensus when the same dataset keeps getting used. Dissorophids, the armoured dissorophoids, are a great example of this. There is a ton of work, but pretty much everyone uses the same phylogenetic matrix. As you might imagine, analyzing pretty much the same dataset a bunch of times should lead to pretty much the same result, which would not be particularly compelling as a "consensus," but, SPOILER, there is in fact no consensus among these nearly identical analyses! That's a huge issue. This study delves into why, both through a separate phylogenetic matrix and from closely scrutinizing the previous one. There is a ton of data packed into it, but the short and skinny are that (1) previous studies have been compromised by systemic errors from the original; and (2) there is a general lack of resolution / of well-supported nodes among dissorophids (i.e. we don't know a lot more than we do know). Then there's a bunch of other toss-ins like missing holotypes and chimeras. Keep reading to find out more!
The limb gets a lot of attention because it's the part of the body in direct contact with the ground (or in the case of some animals, avoiding contact with the ground). But equally important is the apparatus that connects to the limbs - the girdles! What we call the shoulder girdle is scientifically termed the 'pectoral girdle,' while what we call the hip or the pelvis is termed the 'pelvic girdle.' This week's post will look at the pectoral girdle! Temnospondyls have four to five different elements in their pectoral girdle: the interclavicle, the clavicle, the cleithrum, and the scapula + the coracoid / a composite scapulocoracoid. Stem tetrapods (like Acanthostega in the middle below) have an anocleithrum inherited from our fishy forerunners, but this is absent in all temnospondyls. Conversely, some extra bones are found in modern amphibians (like Salamandra on the right below). Several of these will sound unfamiliar to people who only know mammals because we (and other mammals) lack cleithra and interclavicles. 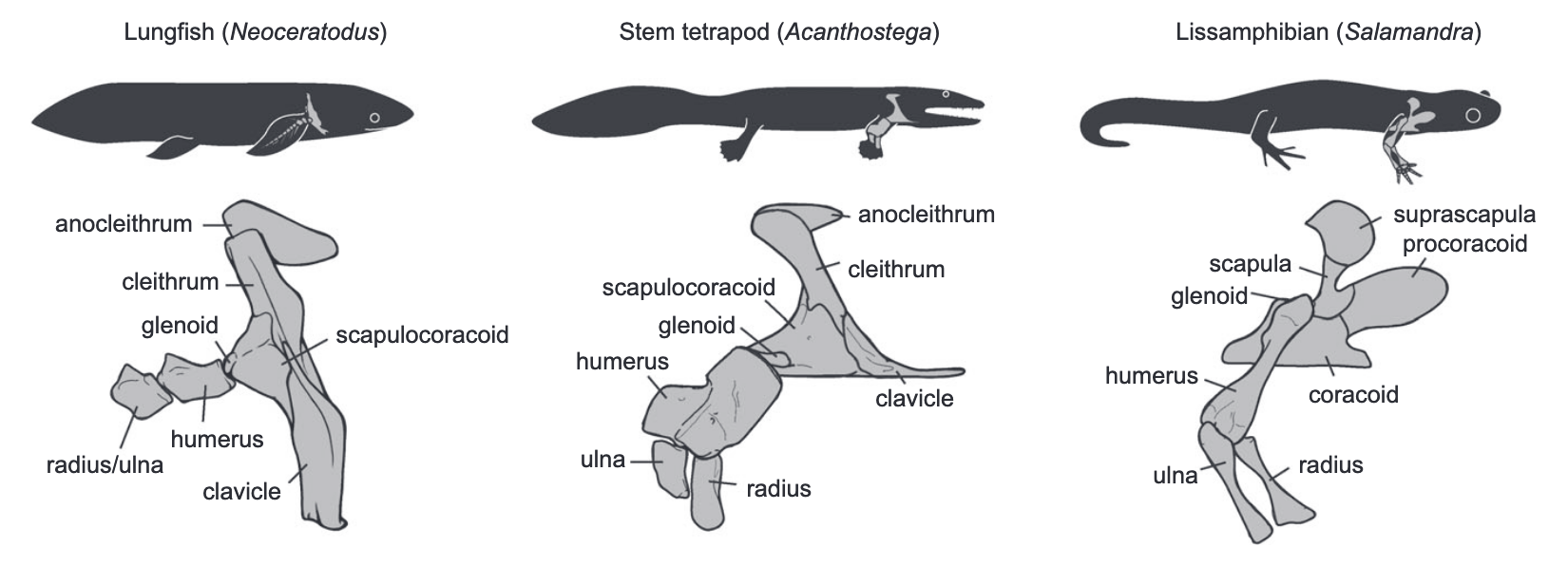 Comparison of the pectoral girdle and upper forelimb in (A) the living lungfish a representative extant sarcopterygian fish Neoceratodus; (B) the stem tetrapod Acanthostega; and the living salamander Salamandra (source: Molnar et al., 2017). |
About the blogA blog on all things temnospondyl written by someone who spends too much time thinking about them. Covers all aspects of temnospondyl paleobiology and ongoing research (not just mine). Categories
All
Archives
January 2024
|
 RSS Feed
RSS Feed
