|
Title: Returning to the roots: resolution, reproducibility, and robusticity in the phylogenetic inference of Dissorophidae (Amphibia: Temnospondyli) Authors: B.M. Gee Journal: PeerJ DOI: 10.7717/peerj.12423 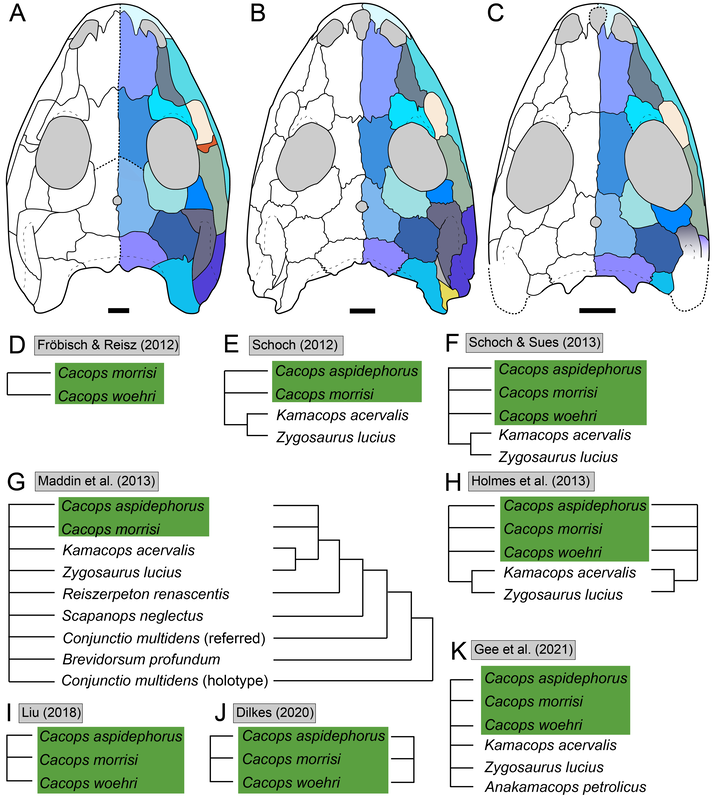 General summary: Phylogenetics, the means of inferring relationships among organisms and one way of classifying them, is a timeless part of paleontology, in part because our datasets are much more limited than those for modern organisms in which genetic barcoding is both widely accessible and robustly informative. As a result, there is often a lot more uncertainty and disparity among different analyses, and people should generally be skeptical when someone says there is a strong consensus for a given group, no matter how well-known (paleo nerds will definitely recall the debate a few years back about what 'Dinosauria' constituted). Temnospondyls are generally not well-known, and this means that they are not as well-studied. This often has the effect of resulting in one or two people producing a lot of the work for a given group, which can lead to an overstated consensus when the same dataset keeps getting used. Dissorophids, the armoured dissorophoids, are a great example of this. There is a ton of work, but pretty much everyone uses the same phylogenetic matrix. As you might imagine, analyzing pretty much the same dataset a bunch of times should lead to pretty much the same result, which would not be particularly compelling as a "consensus," but, SPOILER, there is in fact no consensus among these nearly identical analyses! That's a huge issue. This study delves into why, both through a separate phylogenetic matrix and from closely scrutinizing the previous one. There is a ton of data packed into it, but the short and skinny are that (1) previous studies have been compromised by systemic errors from the original; and (2) there is a general lack of resolution / of well-supported nodes among dissorophids (i.e. we don't know a lot more than we do know). Then there's a bunch of other toss-ins like missing holotypes and chimeras. Keep reading to find out more! This is an example of an admittedly "boring" paleontological study. There are very few pictures of fossils, none of which are particularly nice specimens; the results will never turn into some kind of remarkable paleoartistic reconstruction; and you will never hear about these findings in the news. Nonetheless, studies like this underpin the very foundations of paleontology and all of those attention-grabbing paleontology headlines, which hopefully will become apparent from this post / paper. Phylogenetics 101 Before getting in to the details of this particular paper, I think it's important to give an overview of phylogenetics for readers who maybe don't know very much (or anything) about it. (Those familiar with phylogenetics can keep scrolling). I've touched on phylogenetics before in relation to some of my other papers, but I'll review some key terms and ideas here, beginning with "what is phylogenetics?"
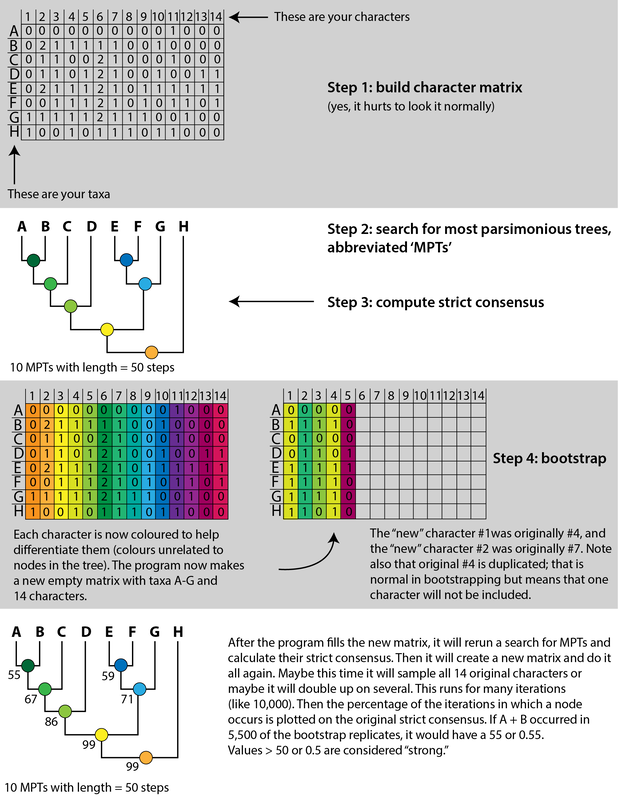
The data. Data entry is the first step in computer-assisted phylogenetics. What we do is make a character matrix with a lot of numbers. The image below is a partial screenshot of one matrix that I worked with for this study. On the left-hand side, you have all the names of the different species being studied. On the top-row, oriented diagonally, you have names of different characters (these are different features being assessed). The rest is made up of numbers, which are colour-coded in this example. 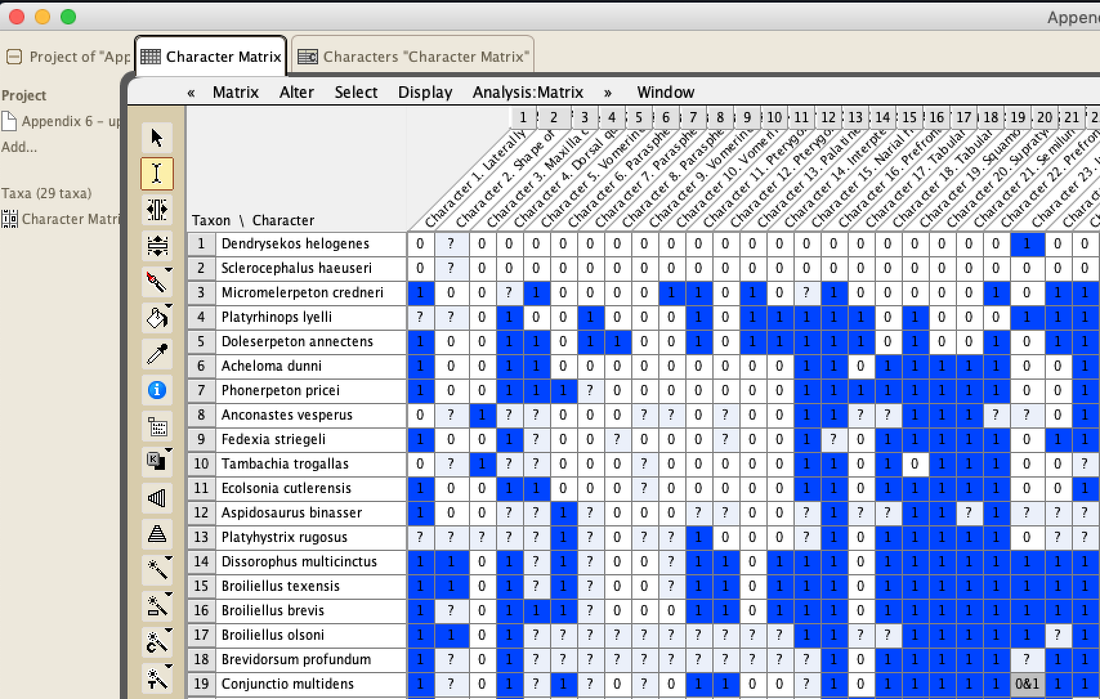 Phylogeneticists begin by sampling taxa - which species do they want to study? Without going into details, paleontologists have particular challenges because some taxa are only represented by fragments, and these tend to be very bad for phylogenetic analyses because they are scored for very few characters, so usually these are omitted. It is very rare to include every species of a group that you are interested in. Then they sample characters - which features differentiate these species? A simple example:
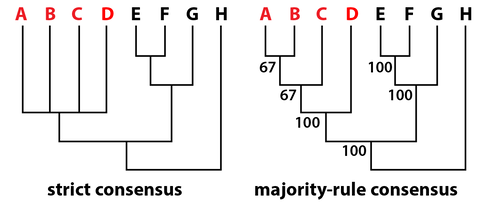
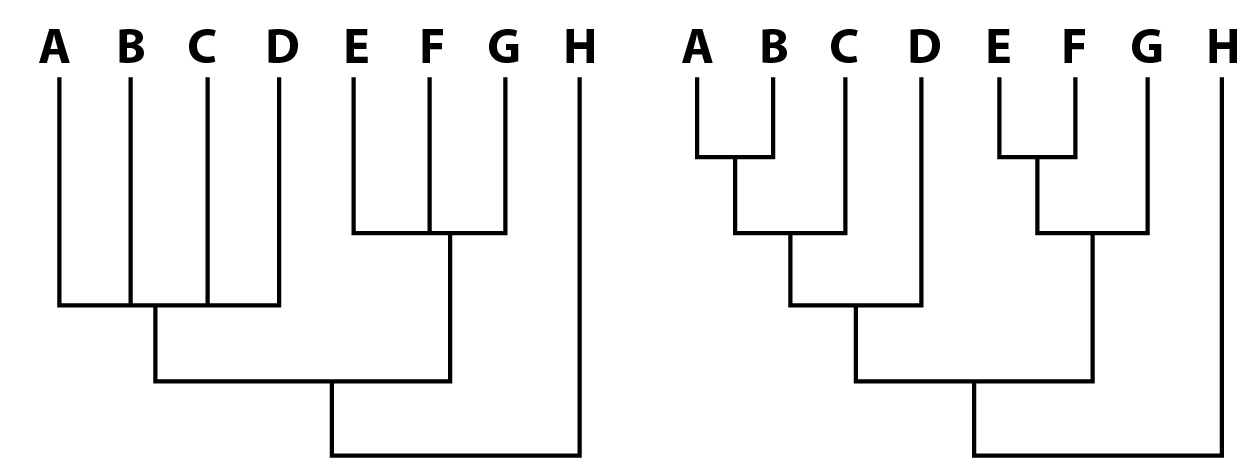
It's important to note that the simplest explanation is not always the right one. Life finds ways to do some really weird stuff. The gist is that parsimony is a workable baseline. Once we allow for more complicated solutions, the number of possible solutions increases exponentially, and it becomes both computationally expensive to search for them and difficult to actually figure out which one is the most likely (but this is where likelihood methods come in, which I'm not talking about here). The output. A parsimony analyses will return what are called the most parsimonious trees (MPTs). There can be as few as one but are often many more. All of these MPTs have the same "length," which is given as a number of steps, which are essentially the number of evolutionary changes that had to occur to produce a tree with these relationships. If there are multiple MPTs, they will differ from each other at least slightly. In the example below, we recovered three MPTs. The red "taxa" (A–D) are ones whose relationships differ between each MPT. Note that some taxa may have the same relationships across all of the MPTs. 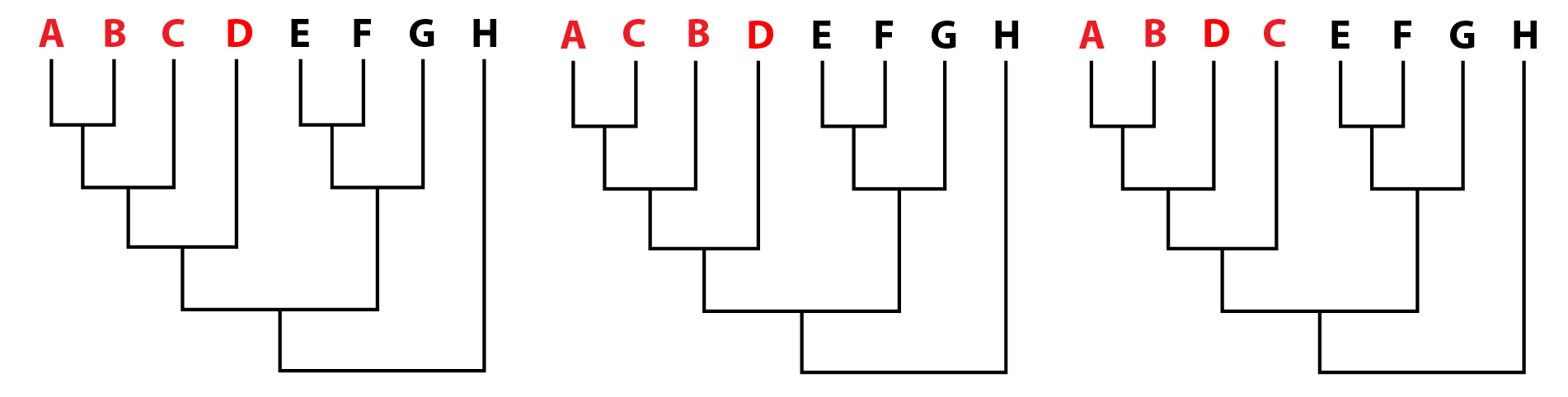 We then have different ways of summarizing the results (some of these are controversial).
 The actual study (finally)The system Okay, we are finally to the part where I talk about the actual study! This study looked at a group of dissorophoid temnospondyls, a group mostly found in the late Carboniferous and early Permian that I worked on for my dissertation (this is not part of my dissertation though). More specifically, I was looking at the large terrestrial dissorophoids, collectively known as olsoniforms (named after E.C. Olson) and which includes two groups, dissorophids and trematopids (yes, 'dissorophid' is one letter away from 'dissorophoid' and requires careful reading to avoid confusing). 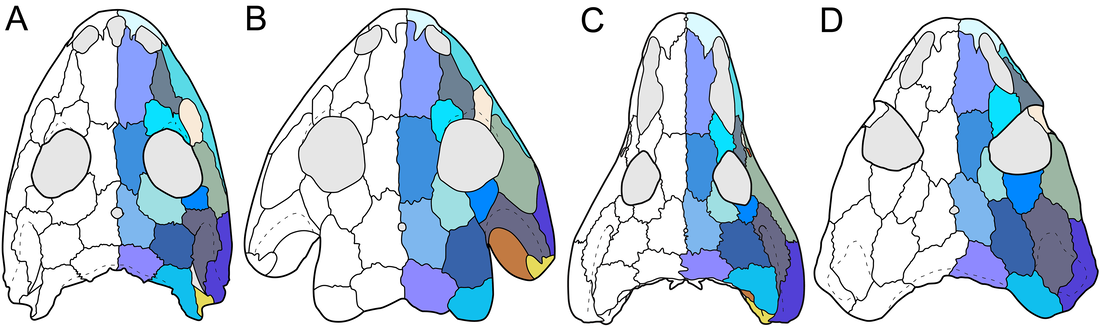
Now what I wanted to do was to take the matrix I made for that 2020 trematopid study and expand it to include dissorophids, thereby forming an olsoniform phylogenetic matrix that comprehensively sampled all appreciably known taxa (no previous study has done this). What I ended up doing was a lot more than that, which is where the alliterative title comes in: "Returning to the roots: resolution, reproducibility, and robusticity in the phylogenetic inference of Dissorophidae (Amphibia: Temnospondyli)." I'll go through these three themes: resolution, reproducibility, and robusticity next. Resolution
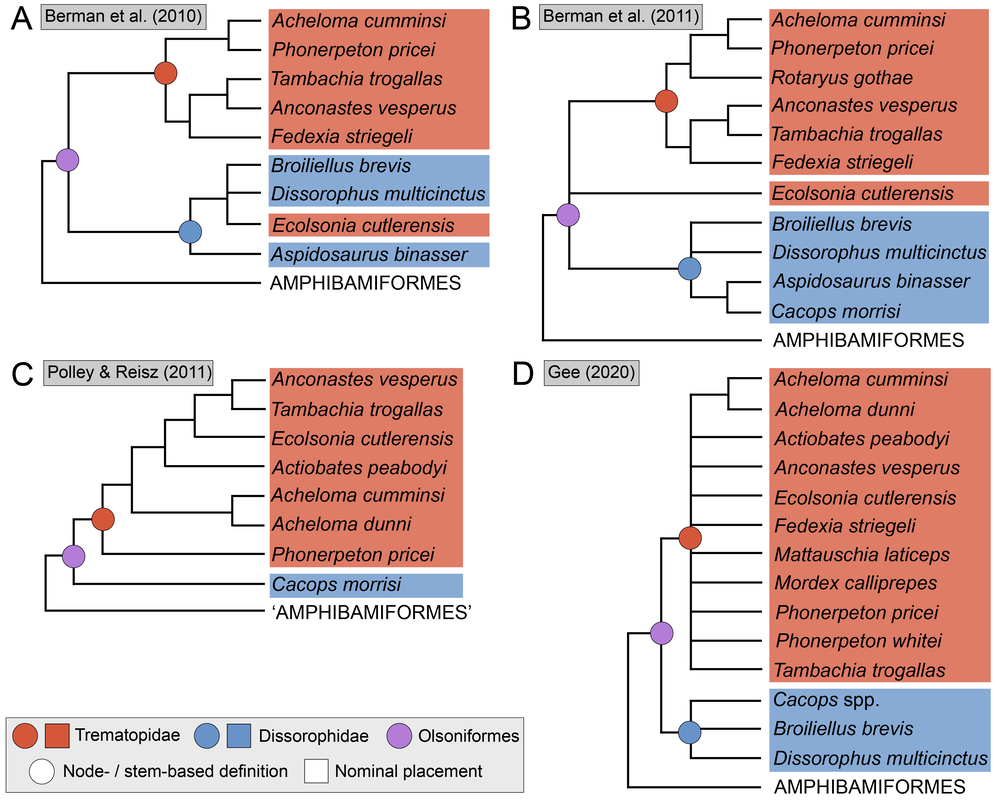
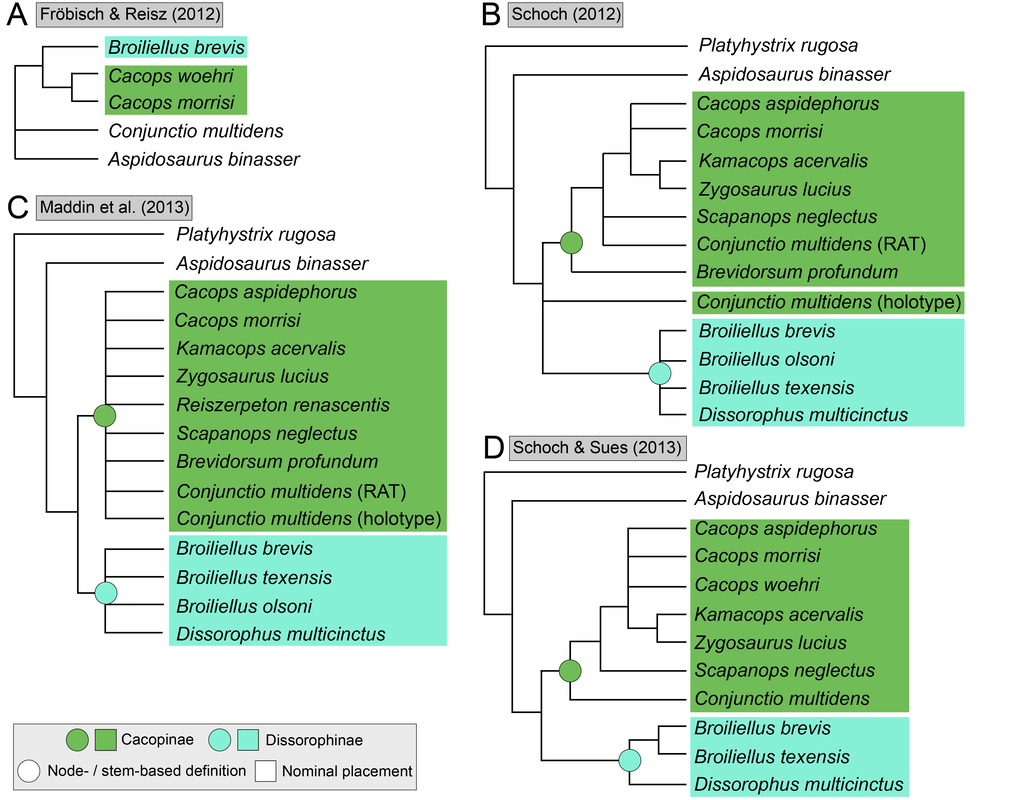
Recent dissorophid phylogenies, shown below, display a range of resolution. There are two points of emphasis here. One is that disparity between studies. Most of these show large polytomies, but Liu (2018) nearly got the fully resolved tree that we're looking for. The second point speaks to what I commented on above - fiddling with the analysis to get resolution where none existed before. If you look at Dilkes (2020), there are two trees. The one on the left is poorly resolved. This is not very helpful for discussing relationships or evolution. The tree on the right is very resolved. We like that one! But to get that tree, Dilkes had to remove four taxa from the analysis. These taxa, often called "wildcards," are problematic in the sense that they confound resolution, whether because they are really fragmentary and have a lot of missing data or present weird combinations of features that don't line up with general trends. Removing wildcards is a common strategy to boost resolution, but like I said above, it inherently omits data that you know exist. 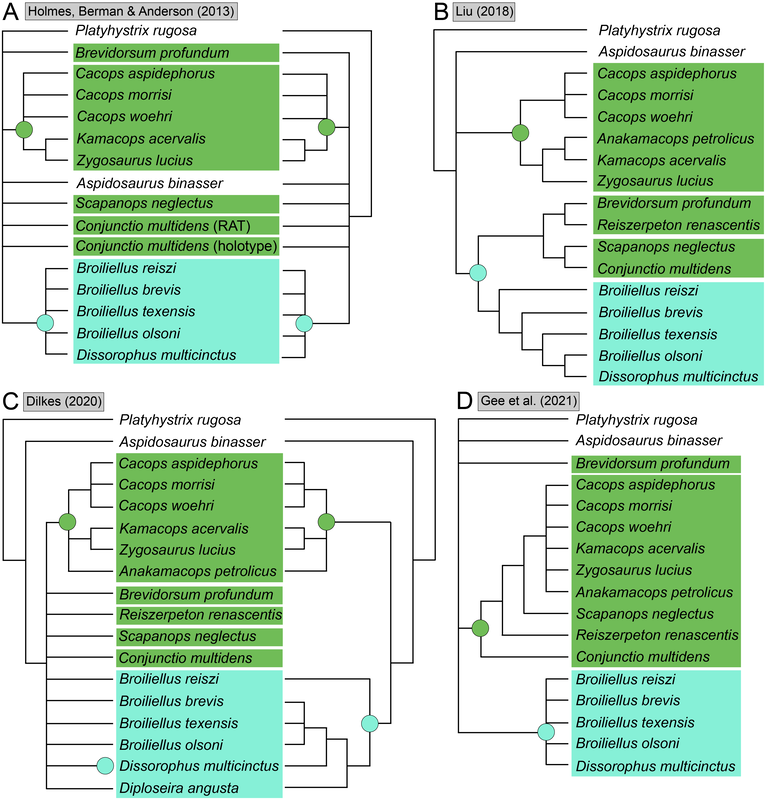 The problem crops up when people use the reduced-data results, which gives the more resolved tree, to talk about evolution and classification schemes, but then place omitted taxa in that framework. For example, Brevidorsum profundum is a typical dissorophid "wildcard." It's only known from one partial skull described in 1964 and is not well-preserved. Some people don't even think it's a dissorophid. Brevidorsum is either recovered within a large polytomy (uninformative) or is excluded from the analysis, but some workers constantly refer to it as being a member of a specific subfamily within dissorophids: Cacopinae (the dark green boxes). However, no recent analysis has ever demonstrated support for that, and this is where the old-school qualitative methods ("it sort of looks like that") and the modern quantitative methods ("the analysis says X") combine in a bad way.
My own analyses tended to recover very poor resolution. Sometimes this could be attributed to including a lot of pretty fragmentary taxa that aren't normally included (like the analysis shown above on left). When it looks like a giant comb...that's an uninformative tree. I could further extend that by showing that removing just a few taxa, some of which are not that badly preserved, could lead to a drastic increase in resolution (like the analysis shown above on right), similar to that example I just cited above. Summary: Resolution is generally quite poor in olsoniform, dissorophid, and trematopid analyses. The only way to get decent resolution that allows one to create a detailed narrative of evolutionary history is to remove a fair number of taxa from the analysis, which only allows you to create a coarse-level narrative that often omits the "weird" taxa that may in fact figure prominently in a clade's evolution. Reproducibility Reproducibility is one of the foundational tenets of science. If someone does an experiment again, can they get the same results? The first step in this process is that it has to be clear how to reproduce an experiment. If you run a complicated study of colour preference in spiders, but only say "we picked some spiders and not others for this experiment" in your methods, it will be impossible for someone to know the specifics of how you did the experiment. So even if someone redoes an experiment on colour preference in spiders, it will likely not be the same as your experiment. Maybe they picked the wrong spiders. Or used different colour palettes. Etc. Because of this, whether their results are the same or not ("spiders like flaming hot pink") as yours, comparing them is difficult because differences in the methods could be responsible for the outcome. In phylogenetics, the main areas where reproducibility comes into play are (1) the character matrix; and (2) the parameters of the analysis. Most journals require people to include their character matrix in one form or another, but ambiguity in the requirements is one problem – some people include their matrices in very inaccessible formats that have to be modified or typeset before a program can analyze them (even though the software to make universally accessible formatted files is F-R-E-E and easily available). In the simplest format, all of those scores in the nice GUI display as text strings:
So when people don't provide their scores in an accessible format like the NEXUS file above, that means the next person has to edit or transcribe the data, which can introduce errors (indeed, I found a few errors in older studies where they had to transcribe it from a previous study and introduced a typo). Below is an example of a very inaccessible data matrix (the dissorophid matrix of Schoch, 2012); this is a figure of a table that cannot be OCR'd (even with the original publisher's version). This means that anyone who wanted to use this matrix would have to type out all of these scores themselves! That is a big waste of time.  Reproducibility doesn't always have to be about repeating the exact same experiment though – more straightforward experiments or ones where the results are intuitive (i.e. doesn't seem like there was data manipulation or accidental error) can be a waste of time to repeat except in coursework. So an alternative way to test reproducibility is to see whether an independent approach to the same question can recover the same solution. If the same general results are recovered, this really strengthens the argument derived from them. If there are different results, that provides an opportunity for future work to figure out why there are discrepancies. 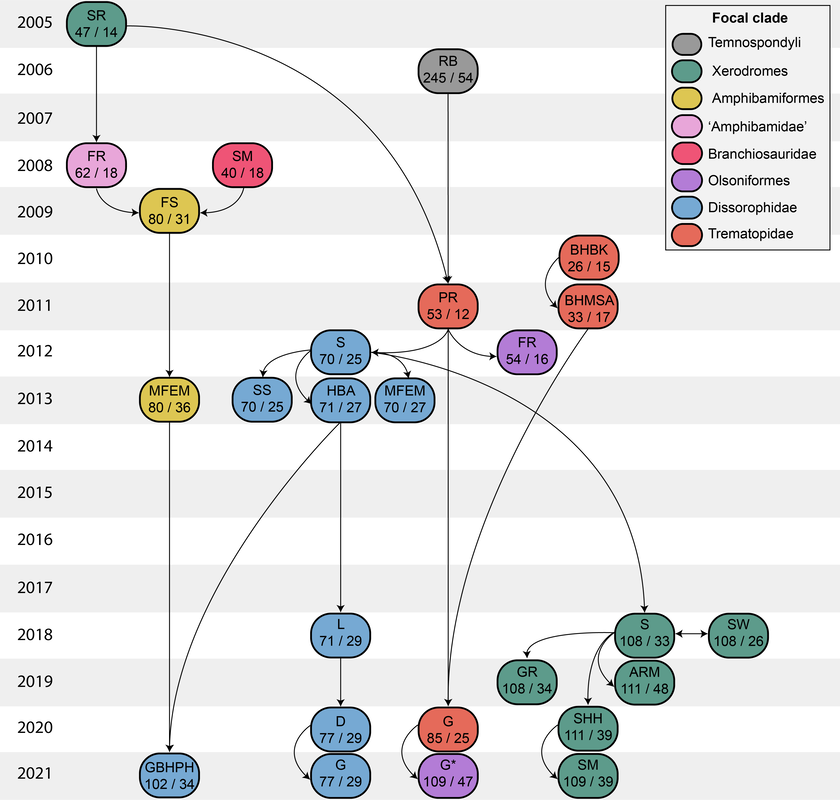 One characteristic of most previous dissorophid studies is that they all draw from the same source matrix (Schoch, 2012; this is the blue bubble in the 2012 row in the figure above). This is common practice in science – taking an existing matrix and adding taxa of interest. It saves a lot of time because creating a new matrix is a lot of work! But there are two important points here. The first is that if everyone uses the same matrix and doesn't make many changes, each derivate of that matrix is not fully independent (we call these pseudoreplicates). Each pseudoreplicate is predisposed to producing the same results as the last iteration because the underlying data are nearly the same. So getting the same general result from five pseudoreplicates is not as much of a "consensus" as getting the same general result from five independent matrices. Essentially, this can be misleading and make you think that the results are very reproducible when that is really just because the data are nearly identical. This leads to the second point, which is that the data must be solid when they're getting propagated, otherwise you're just propagating errors. Immediately, there is a big red flag when looking at dissorophid phylogeny:
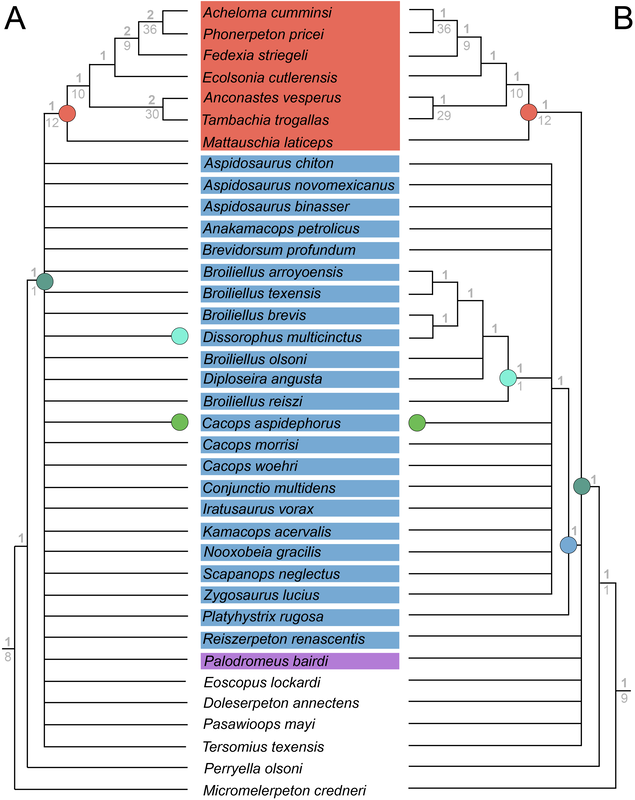
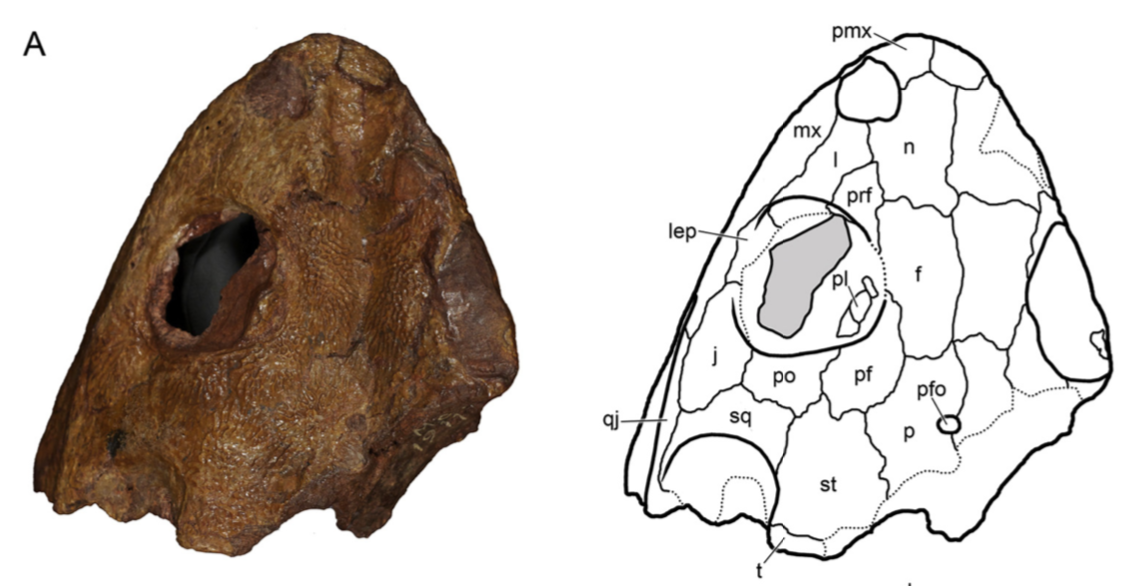
There is no consensus among these pseudoreplicates, in spite of being 99% similar to each other!. Not getting a consensus from a bunch of pseudoreplicates is really concerning because it indicates that relationships are not well-supported and can easily change with just the addition of one taxon or a few scoring changes! The explanations. There are a lot of different explanators for why there are different results. Some of these are a little more towards personal preference in setting different parameters in the analysis. One probable explanation is that the taxon sample differs slightly between studies; those with more resolution tend to have omitted more of those "wildcards" that are weird or highly fragmentary. If you compare the results of Maddin et al. (part C above) and Schoch & Sues (part D above), this is a likely explanator for why the former is much less resolved - it includes historical wildcards like Brevidorsum profundum, fragmentary taxa like Broiliellus olsoni, and somewhat odd taxa like Reiszerpeton renascentis (the holotype of which was long considered to be an amphibamiform dissorophoid). Taxon samples are low-hanging fruit for explaining topological differences. So I went deeper and started looking at specific cells in the matrix to see if there were perhaps typographic errors (I did find some of those). However, I found a lot more errors that do not seem to be typographic. I looked at the latest version of the Schoch (2012) matrix, which was the one published by Dilkes (2020). I identified over 140 scoring errors, which is a lot in a fossil matrix that only has up to 77 characters and 29 taxa. Many of these were instances where Taxon X was scored for Feature Y...except Feature Y isn't even known in Taxon X! One example is Cacops woehri from the Richards Spur locality. This taxon is only known from partial skulls, but somehow it was scored for a bunch of postcranial characters. Suspiciously, all 14 scores of this taxon that were erroneous were scored the same as Cacops morrisi, also from Richards Spur but which is fairly distinct from C. woehri. In fact, this deep dive revealed that the three species of Cacops, which are readily differentiated from each other, didn't actually differ in any scores other than their distribution of missing data (i.e. when they could be scored, they were all scored the same). This explains why all previous analyses recovered them as a single polytomy, when qualitative comparisons clearly show that C. morrisi and C. aspidephorus are much more similar to each other than to C. woehri (which might not even be Cacops). Cacops was the most egregious example of what seems to be previous workers "assuming" scores that are not validated by the present fossil record, but other taxa also seemed to have scores here and there where a score was "assumed" based on the general affinity of that taxon (e.g., that some dissorophids with no known quadrate had a dorsal quadrate process preserved; this is a common feature in dissorophoids). This is, of course, a major problem! If you have what you think is a new species of Cacops, and you score it for a bunch of features of other species of Cacops, you have pretty much guaranteed yourself of the outcome that you are allegedly testing because you have made this new species more similar to its alleged relatives than the fossil record actually bears out. Cacops aspidephorus is the best example of this - for over a century, nobody knew what the sutures were because the specimens were so poorly preserved. You could see this if you looked at reconstructions of the skull of different dissorophids, like from Schoch (2012) below. As you can see, C. aspidephorus is just a grey blob - no sutures. Why then, were there 15 scores for C. aspidephorus for characters that require sutures to be known? The sutures of the skull, based on a few specimens that were not so bad off, would not be published for another eight years until Anderson et al. (2020). In fact, other than characters with missing data for one or two of the three species, there were no characters for which one species of Cacops was scored differently than another. 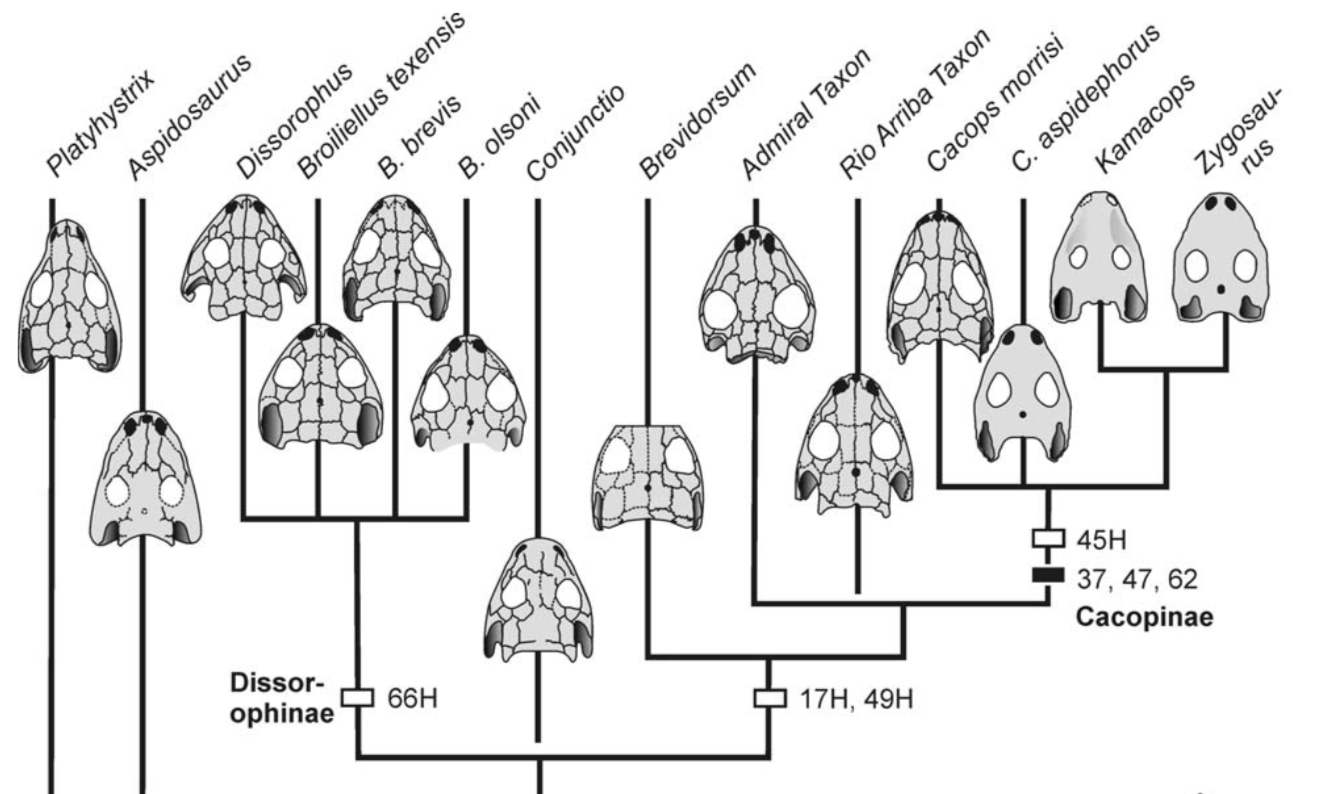 Other issues relate to how characters are defined. For example, if a character requires skull length to be known, it doesn't make sense to score a taxon only known from specimens that aren't complete longitudinally. However, taxa like these were scored for these characters. Issues with characters like these that explicitly require certain landmarks but that seem to be scored in the absence of at least one were fairly common as well. In the examples below, both Brevidorsum profundum (only one specimen; Carroll, 1964) and Reiszerpeton renascentis (only one specimen; Maddin et al., 2013) are known from incomplete specimens. Therefore, their skull length is not actually known, but characters that require a ratio invoking the skull length were scored for these taxa previously.
As one might expect, correcting a large number of errors and then rerunning an analysis leads to drastically different results. On the left of the figure below are the original results from the Dilkes' matrix, and on the right are the results when the corrected version of the matrix is analyzed. There is a major loss of resolution! This suggests that at least some of the inconsistencies between previous pseudoreplicates of this matrix result entirely from erroneous scores in the matrix. 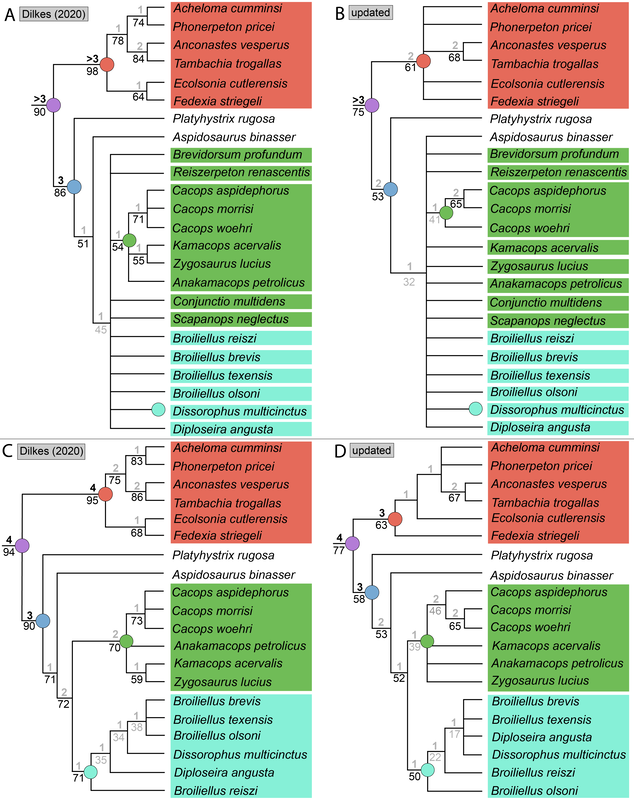 Furthering the issue is that these systemic errors didn't just appear in the latest version of Schoch (2012), published by Dilkes (2020). In fact, none of them were introduced by Dilkes, which I feel is very important to state, lest the reader think that Dr. Dilkes produces error-riddled work. Most of the errors go back to the original matrix and were thus introduced by Schoch, although some were separately introduced as taxa were added (e.g., Holmes et al., 2013; Liu, 2018). This means that every single previous study that used this matrix has also been compromised. I suspect, though I didn't test this, that correcting the same scores in all of the previous matrices would result in a loss of resolution in all of them, but this would actually boost the similarity between studies because the resolution of wildcard nodes would be eliminated. Summary: Previous dissorophid analyses are highly irreproducible insofar as the source matrix (Schoch, 2012) contains substantial errors, many of which seem to be based on assumed anatomy that cannot be validated from the fossil record. This has likely compromised studies that analyzed this matrix or a derivate thereof with respect to resolved relationships within Dissorophidae (unresolved relationships are probably okay). Robusticity Without some assessment of reliability, a phylogeny has limited value. It may still function as an efficient summary of available information on character-state distributions among taxa [...] but it is effectively mute on the evolutionary history of those taxa (Sanderson, 1995:299). One of the challenges with strictly morphological data is that there are just not very many datapoints. Compared to thousands to millions of genetic base pairs in molecular analyses, the average fossil tetrapod matrix has between 50 and 350 characters. This means that just a few scores could be responsible for a certain recovered relationship, whether a longstanding one or an entirely new one. Sometimes, even changing just a single score can change the entire tree! This is where support metrics come in. These are statistical means of assessing how "strong" or "robust" a given node is; in other words, is this relationship likely supported by only a single score, and thus not very reliable, or are there many lines of evidence (scores) supporting it? In parsimony analyses, there are two conventional methods of assessing support. Bootstrapping: Say you have a character matrix with 8 taxa and 14 characters. Bootstrapping is called "resampling with replacement." In one bootstrap replicate, the program will randomly select a character and its correspondent scores. Let's say it selects character #7 from the original matrix. This now becomes the new character #1 in the bootstrap. The program then picks the new #2 and the new #3 and so on. The caveat is that it can pick a character from the original matrix that was already selected. So it could pick character #7 three times in a row. It keeps picking until it has the same number of characters as the original matrix. Invariably, it is almost a given that some characters will be resampled multiple times, which means others are not sampled at all. Then you run this a lot of times (like 10,000) and calculate the percentage of these resampling replicates that nodes are found in. If you get low bootstrap support, that means very few replicates recovered that relationship and suggests that it is not well-supported because it is probably linked to one character. Bootstrap support over 50% (a node occurs in >50% of the bootstrap replicates) is considered "strong."
 Bremer decay index: Bremer support is an entirely different measure that has nothing to do with resampling. Bremer decay calculates how many additional "steps" are needed for a given node to collapse. This is exclusive to parsimony analyses as a result since it is predicated on the number of steps in the most parsimonious tree(s). Some programs have an automated way of calculating Bremer support, but the common way to do it is to get the MPTs and the number of steps among them, then get the strict consensus. Then you rerun the search and tell it to save not only the MPTs but also all trees that are +1 step longer. This will inherently be more trees and it includes trees that are not the most parsimonious. Then you get the results, compute the strict consensus, and if a node previously found is no longer found, that node has "collapsed." If it collapses at +1 steps, then its Bremer decay index is 1. You then proceed to do this search for all trees +2 steps longer, +3 steps longer, etc. >3 is considered "strong." 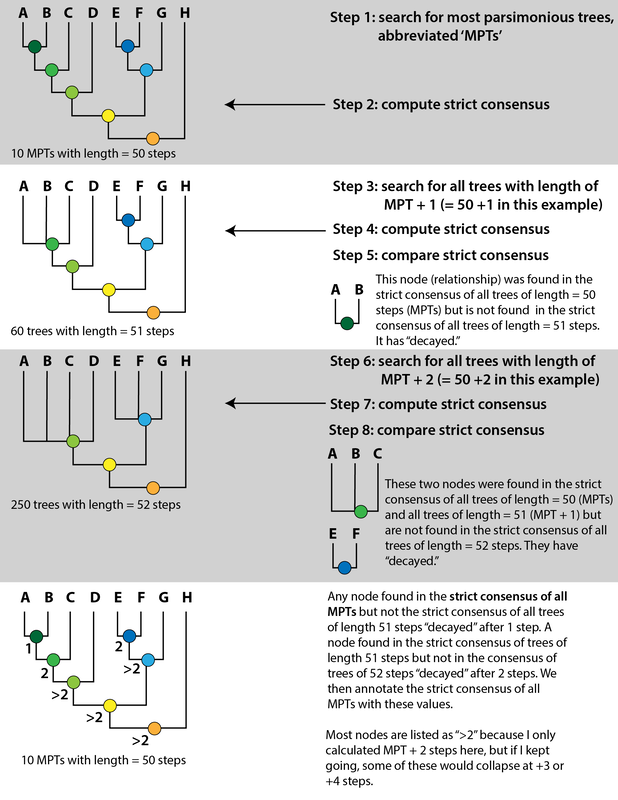 It is standard for scientists to report at least one if not both of these support metrics because it tells you how robust your recovered tree is. If a tree is not well-supported, then even one scoring error or dubious score could be producing this topology. Analyses with my matrix tended to recover low support for at least one of these metrics. I used a visual key to indicate whether a node was strongly supported or not by marking "weak" support in greyed-out text. You can see in the example below, which was an analysis of my matrix with the same taxon sample as Dilkes (2020), that there is a lot of grey... In fact, in this particular analysis, the only node with both strong Bremer and bootstrap support is the node for trematopids (orange circle), which was not even a focus of this analysis! 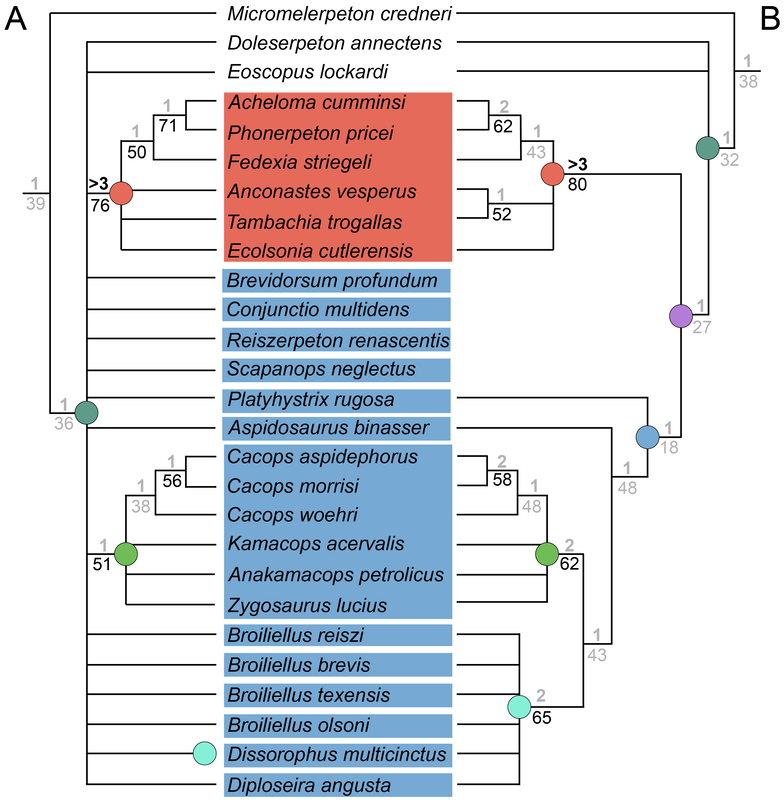 Previous dissorophid studies are a giant mixed bag. Some studies only reported one metric, and others didn't report anything. The latter is particularly bad because it emphasizes getting resolution (intentionally or not), no matter how weakly it might be supported or how few characters it might hinge on. This can lead people to "tinker" with the matrix, leaning one way when they're on the fence or specifically making modifications to certain characters or taxa until the expected or desired result comes out. It might be a reason why there were so many errors in the Schoch (2012) matrix in which one species was scored the same as another species in the same genus – scoring based on assumptions like this is a great way to boost both resolution and support. It's a reminder that even an "objective" quantitative method like computer-assisted phylogenetics is still underlain by subjective human decisions. Furthermore, when support is reported, it's often weak except for really large clades. For example, it isn't really that surprising or informative in a dissorophid-focused study that Dissorophidae is a strongly supported node unless you were suspicious about whether all dissorophids actually formed a clade (this isn't particularly controversial). 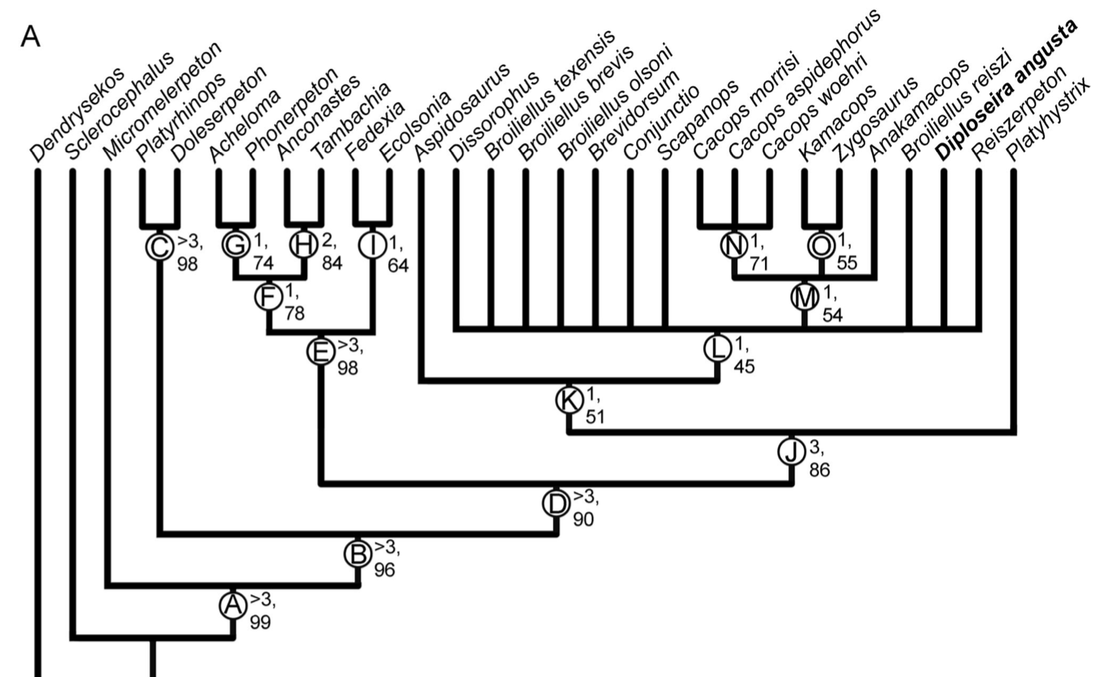 This is an example from the Dilkes (2020) analysis. As a reminder, "strong" support is Bremer > 3 (the first value) and bootstrap > 50 (the second value). Values near the bottom (more inclusive clades) are high, but they get really low as we go up the tree. Like my own analyses, trematopids (node E) have excellent support...but we're not studying trematopids in this analysis... Once you get into Dissorophidae (node J), which is what we wanted to study, no in-group node has Bremer > 1 (literally the lowest it can be) and most of them have bootstrap hovering around that 50% threshold. This isn't an indictment of the scientist, just to be really clear. But it does underscore that the topology of the tree is not the only thing that matters! Weakly supported relationships are just that – weakly supported. Summary: Even when there is resolution achieved in dissorophid analyses, the statistical support is weak for most nodes except those that are outside of Dissorophidae. So this suggests that the inconsistent topologies between pseudoreplicates of Schoch (2012) are all insignificant insofar as one is not preferable because none is strongly supported statistically. Reporting these stats is important, even if it hinders what author(s) can say. A few other goodies... Having already gone way further into the weeds than I intended to for this project, a lot of the discussion is a comprehensive synthesis of where things stand with dissorophids in their entirety. I overview Cacops, which could get a little help from a revision of Parioxys, a taxon originally interpreted as an eryopoid (and thus commonly placed as one in supertrees) but that in fact seems to share many similarities with Cacops. It has not usually been compared with dissorophoids at all, but could it be one? I also commented on Broiliellus, which is a dumpster fire of a genus. Nobody has ever recovered Broiliellus as a clade except by...you guessed it...excluding taxa. My preference is to restrict Broiliellus to the type species, B. texensis, and put everything else in single quotes (e.g., 'B.' reiszi) until this can be sorted out (some of these taxa have not been described since the 60s). 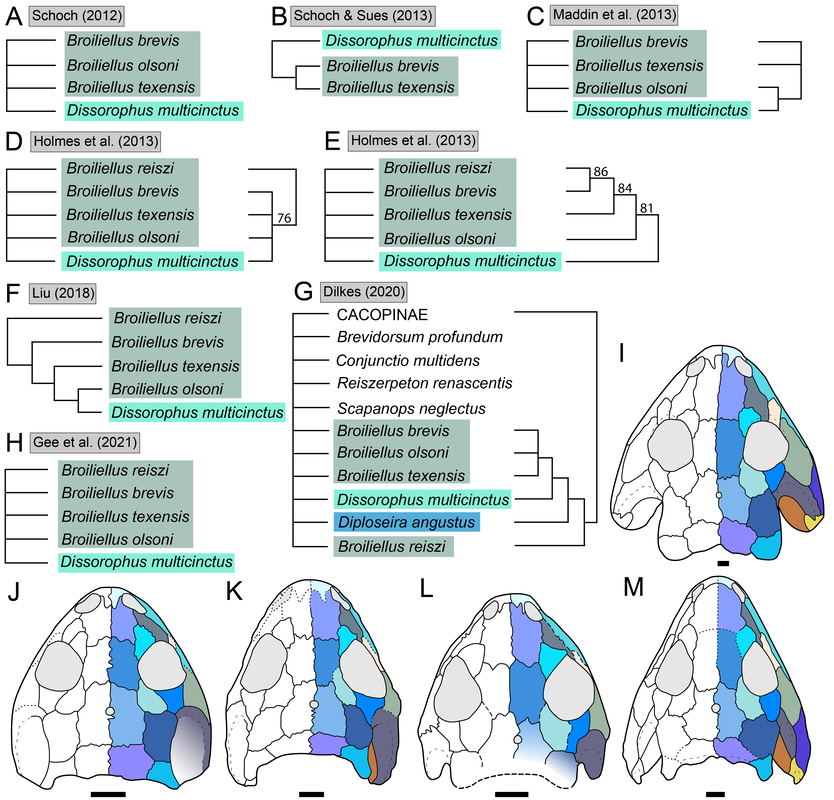 Then we have two more exciting discussions, the first a missing holotype and the second a possible chimera.
Allegedly, AMNH FARB 4785 should not exist! Dating back to the 60's, people said this specimen was missing. But I found it! (technically I found it in 2017 and said as much in a 2018 paper). But then where is AMNH FARB 4785a, which was apparently confused for AMNH FARB 4785? Concerningly, that specimen is nowhere to be found, and there is no record of it in the museum database or in their most recent inventory. It is always bad when a specimen goes missing, let alone a holotype. However, as far as I know, this specimen has never even been figured, only described generically as spines. It certainly wouldn't be the first time that a specimen just disappeared.
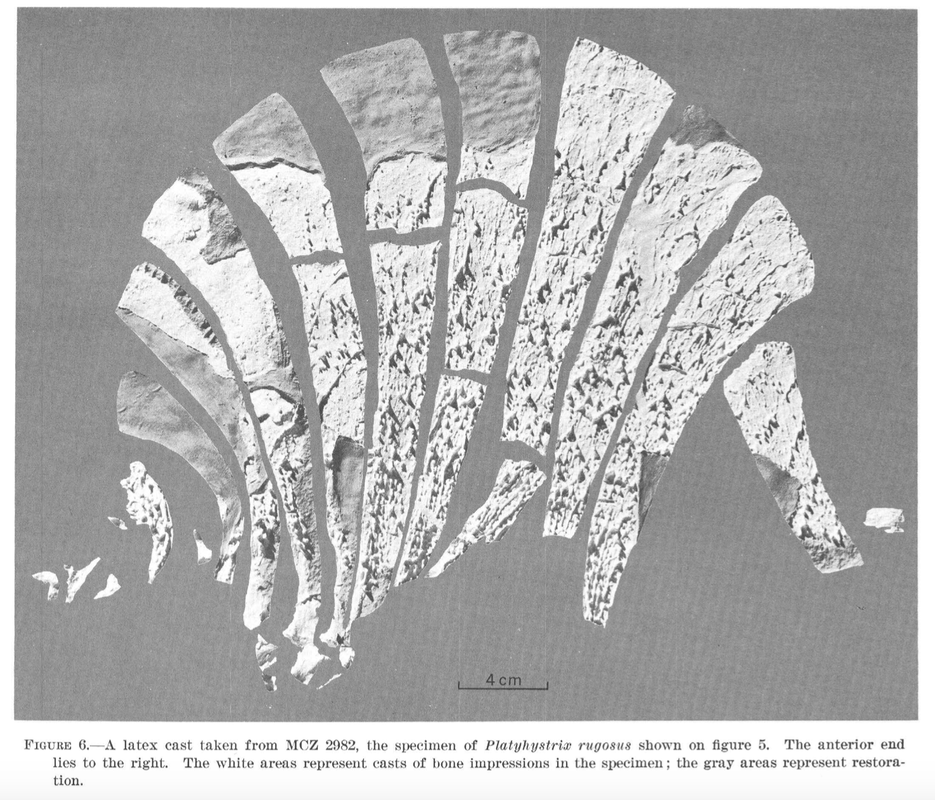
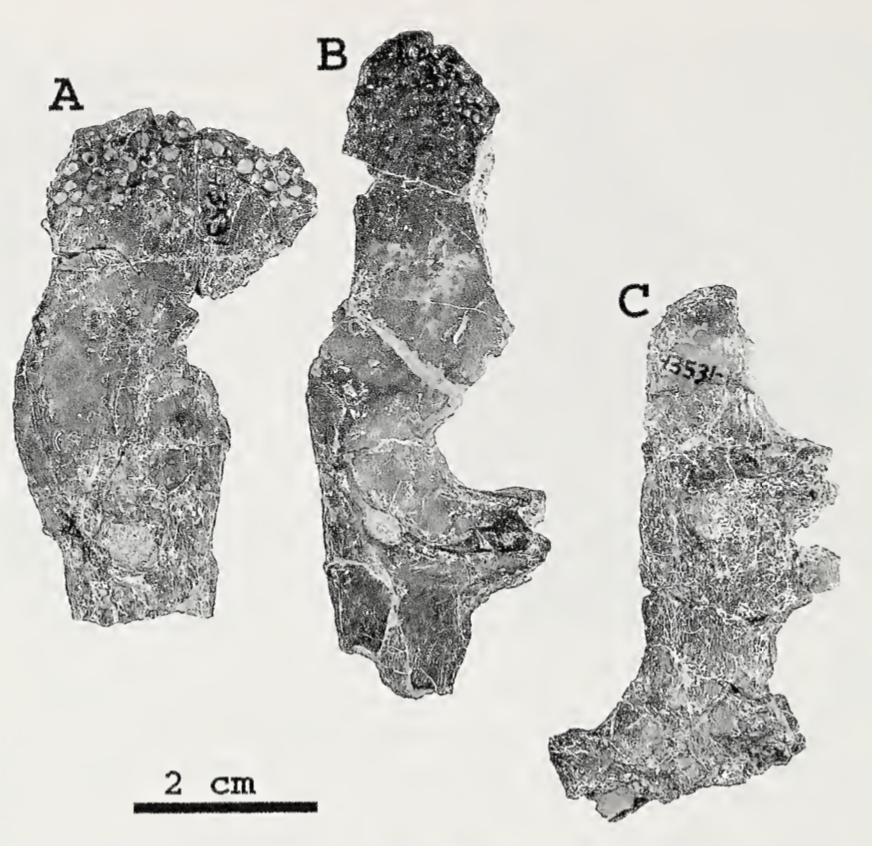
Above is AMNH FARB 11544. These are characteristic spines of Platyhystrix rugosa (part A). Coincidentally, this specimen was collected in 1881, the same year that AMNH FARB 4785a was collected, and they were both collected by the same guy from the same bonebed in New Mexico. However, AMNH FARB 11544 was not described or figured for a century until Berman et al. (1981). However, it meets the original description of AMNH FARB 4785 ("several spines") by E.C. Case in 1910, includes several that are in line with the measurements given by Case, and even includes a partial synapsid scapulocoracoid that was also mentioned by Case. The coincidence is almost too compelling! But there is no record of a number transfer, even after I checked with the AMNH collections staff (who have no record of AMNH FARB 4785a in their database on in the most recent inventory), so at present, the holotype of P. rugosa remains the missing AMNH FARB 4785a. I put this in the paper in the hopes that someone will be able to shed more insight on this... Chimeras
Most examples of chimeras in paleontology are unintentional. They frequently result from assumptions that a collection of bones from the same spot, without excessive elements (e.g., five femora), belong to not only the same species but to the same individual. This is where preconceived biases can come in; everyone will have a list of "likely candidates" for a fossil when they come across it (e.g., you do not expect to find any temnospondyls in the Cretaceous Hell Creek Formation alongside Triceratops), but sometimes this list is too restrictive. A recent example is Dakotaraptor, a putative dromaeosaurid dinosaur (Velociraptor group) in which the purported wishbone turned out to be part of a turtle (see here). Another was a relatively rare report of a pterosaur pelvis from Canada that turned out to be part of a tyrannosaurid skull (see here and here). As far as I know, nobody's ever definitively identified a temnospondyl chimera. That's where Aspidosaur binasser comes in. This taxon is based on one specimen from the Permian of Texas. It includes several skull fragments that collectively fill out most of the top of the skull and about 20 vertebral positions. This species is interesting for a couple of reasons. The first is that Aspidosaurus (like "Aspidosaurus" apicalis above) is a wastebasket taxon, encompassing a bunch of generally similar morphotypes that probably don't represent a single taxon. These morphotypes are largely isolated osteoderms that form an inverted V shape; cranial material of Aspidosaurus is practically unknown, which wasn't helped by the destruction of all material of the type species (which did include a skull), A. chiton, during an Allied bombing of Germany in WWII. So A. binasser is the best (and only) skull material we have. Then, A. binasser was described as having three different osteoderm types (shown below; figure from Berman & Lucas, 2003), which is unprecedented variation – other dissorophids only have one type, though it may change slightly in width or other minor attributes. And this is really where things start to get a little suspicious.
Despite having a good portion of the skull, including the occiput, which is the neck joint, and 20 vertebral positions from specifically the presacral (anterior to the pelvis) region, there is no articulation between anything. All three osteoderm types are found on their own; there is no block or fragment with two or three different osteoderm morphotypes on it. Then there is the peculiar question of where are the atlas and axis (the neck vertebrae)? Most dissorophids seem to have between 20 and 25 presacral vertebrae, so what are the odds of getting half the skull and 20 presacral vertebrae but not the neck vertebrae? If this holotype is a chimera, it would be really hard to prove because it's not like some composite Chinese bird that was Frankenstein'd together from different slabs, and all of the material does appear to be genuinely dissorophid in nature. And unlike how finding previously unidentified fits can positively prove association, not finding fits is not necessarily evidence for chimerism.
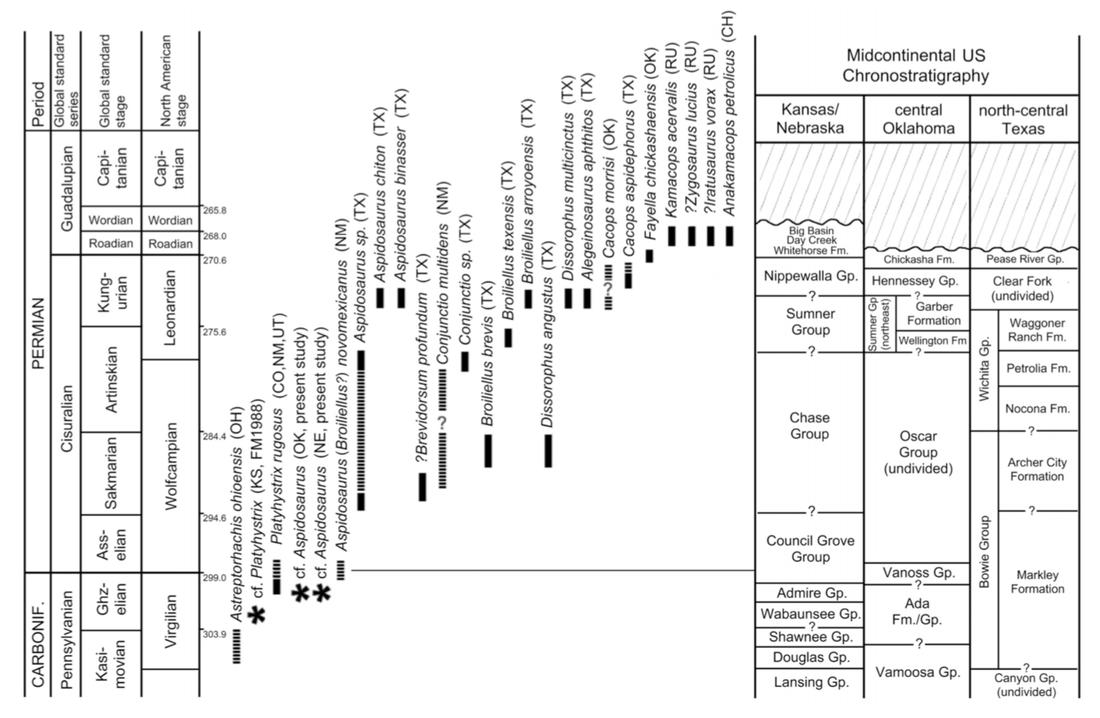 Original caption: Revised stratigraphic distributions of dissorophids in the U.S. midcontinent. Asterisks represent newly reported occurrences from the Pennsylvanian of the northern midcontinent. Solid bars represent observed occurrences. Dashed bars represent inferred or approximated occurrences where specimen provenance is imprecisely constrained or poorly documented. Abbreviations: CH, China; CO, Colorado; KS, Kansas; NE, Nebraska; NM, New Mexico; OH, Ohio; OK, Oklahoma; RU, Russia; TX, Texas; UT, Utah. (Stratigraphic ranges and midcontinental chronostratigraphy were compiled and updated from the following sources: Carroll, 1964; Vaughn, 1971; Olson, 1972; Ossian, 1974; Lewis and Vaughn, 1975; Foreman and Martin, 1988; Hentz, 1988; Hook, 1989; Sumida et al., 1999; Kissel and Lehman, 2002; Reisz et al., 2009; Berman et al., 2010; Lucas et al., 2010.) The original argument (more like assumption) for associating all of this material to a single species, let alone to a single individual, was that there was "no evidence for more than one dissorophid." This is, of course, negative evidence. Just because you don't find five femora doesn't mean that the two that you did find belonged to the same animal or species. Positive evidence would be definitive articulation. At the time, the recognized dissorophid diversity was much lower than it is now – we've had at least five new species named since then – and the interpretation that dissorophid-bearing sites only preserved one dissorophid species seemed to hold up (like with the Cacops Bone Bed). Since then, numerous examples of sites with multiple dissorophids have emerged. Especially at Richards Spur, we know that not only are there many species, but they are not equally abundant or represented by the same elements (more on this). So finding a cornucopia of dissorophid elements does not mean they all belong to one species or to one individual. Aspidosaurus is already known to be known almost only from isolated osteoderms (the same is true of Platyhystrix), so it is not unreasonable to argue that you could have some stereotypical Aspidosaurus osteoderms, possibly with an Aspidosaurus skull, mixed with random bits of other dissorophids. So it is my suspicion that Aspidosaurus binasser is a chimera, but given the nature of its preservation, it is basically impossible to prove. Even if you found a specimen with the same skull that was articulated with vertebrae that only had one osteoderm morphotype, someone could (and probably would) argue that it's just a different species. This is a great example of the burden of dodgy taxonomy – it is very hard to undo (inertia), no matter how weak the original working assumptions were / are. At present, this is not a huge issue for phylogeny because there is no character for number of osteoderm morphotypes, but it is an issue for taxonomy because A. binasser and the functionally lost A. chiton are only differentiated on this feature. Apples and oranges 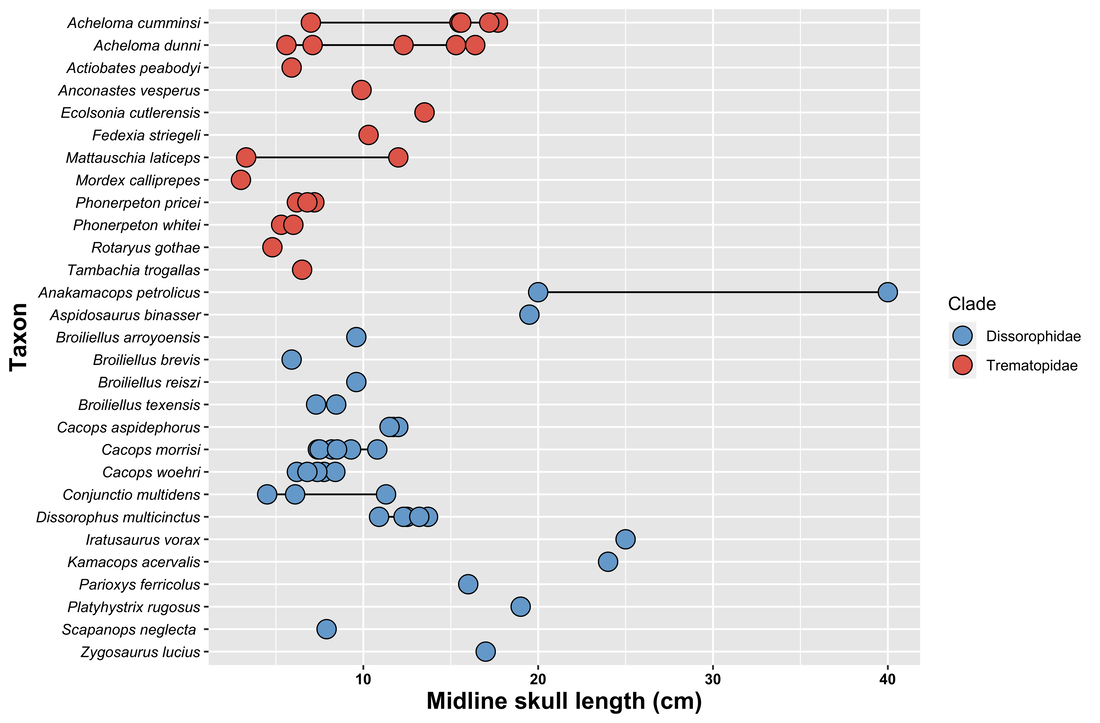 Those who keep up with enough of my research know that one of my big themes is ontogeny, how we identify how mature temnospondyl specimens were at the time of death, and what not accounting for biases or skews in the fossil record might totally screw up our interpretations and analyses. I didn't focus on this too much with dissorophids as I did previously with trematopids, but there is a major disparity in size, which might correlate with disparity in the maturity of preserved specimens. Part of my thesis looked at Cacops and suggested, based on braincase morphology, that pretty much all known specimens of Cacops are juveniles at best, which underscores the point that just because you have a lot of the same size doesn't mean that was the largest size. This is definitely an interesting future direction though because cacopines like Cacops include the largest dissorophoids, which seem to get especially large in the middle Permian after lots of other Paleozoic temnos go extinct (including trematopids). Dissorophids are really diverse, and unlike trematopids, seem to have coexisted with other dissorophid species fairly commonly, so there could be legitimate major size differences between taxa that would lead to niche partitioning (e.g., one targets small tetrapods, one targets medium tetrapods, etc.). Grand summaryThis has been a longggg post. Hopefully it made sense if you read it all the way through, and if you did, thanks for reading! Hopefully you learned something new! This paper can really be distilled to four main points, which I also summarized at the end of the paper in a very atypical fashion for me just because of how much is packed in here.
David Marjanović
11/11/2021 12:21:14 pm
An important paper, and an important post!
Nick Gardner
11/11/2021 08:49:28 pm
This paper is the kind of brutal honesty we need in morphological phylogenetics. Comments are closed.
|
About the blogA blog on all things temnospondyl written by someone who spends too much time thinking about them. Covers all aspects of temnospondyl paleobiology and ongoing research (not just mine). Categories
All
Archives
January 2024
|







 RSS Feed
RSS Feed
